
Fujitsu y la Universidad de Osaka diseñan tecnologías para el cálculo energético de materiales químicos en ordenadores cuánticos
La multinacional tecnológica Fujitsu y el Centro de Información Cuántica y Biología Cuántica de la Universidad de Osaka han hecho público el desarrollo de una nueva tecnología diseñada para acelerar la aplicación industrial de los ordenadores cuánticos en la era de la computación cuántica tolerante a fallos en fase temprana, early-FTQC.
Al combinar la versión 3 de la arquitectura STAR, una arquitectura de computación cuántica con puertas de rotación de fase altamente eficiente y única, con una novedosa técnica de optimización de modelos moleculares, los investigadores han logrado reducir significativamente los requisitos de recursos computacionales.
Este avance permitirá realizar cálculos energéticos para el diseño de materiales químicos, como moléculas catalizadoras, en un plazo de tiempo realista utilizando ordenadores cuánticos early-FTQC. Este tipo de cálculos no es posible actualmente con los ordenadores convencionales y requeriría milenios incluso con versiones anteriores de la arquitectura STAR. Se espera que estas tecnologías contribuyan a abordar diversos desafíos sociales, como la aceleración del descubrimiento de fármacos, la mejora de la eficiencia en los procesos de síntesis de amoníaco y el avance en las tecnologías de reciclaje de carbono.
Antecedentes
La computación cuántica presenta un gran potencial en una amplia variedad de sectores, como el descubrimiento de fármacos, la criptografía y las finanzas. Sin embargo, los sistemas cuánticos actuales presentan altos niveles de error, y se considera que las aplicaciones prácticas requerirán, en general, ordenadores cuánticos con millones de qubits.
Con el objetivo de mejorar la corrección de errores y acelerar la aplicación práctica de la computación cuántica, Fujitsu y la Universidad de Osaka establecieron la arquitectura STAR versión 1 el 23 de marzo de 2023, seguida de la versión 2 el 28 de agosto de 2024. Esta última, equipada con avanzadas puertas de rotación de fase, amplió significativamente la escala de computación, permitiendo potencialmente cálculos early-FTQC de propiedades de materiales en estado sólido, como la superconductividad a altas temperaturas.
No obstante, el cálculo preciso de energías químicas moleculares complejas para aplicaciones prácticas seguía requiriendo recursos excesivos, y los métodos anteriores estaban limitados por una capacidad computacional insuficiente o por plazos de ejecución poco realistas.
Tecnología desarrollada recientemente
Esta investigación conjunta ha demostrado que la combinación de las dos tecnologías siguientes permite realizar cálculos energéticos de materiales químicos con la precisión necesaria y en un tiempo práctico:
1. Desarrollo de la arquitectura STAR versión 3
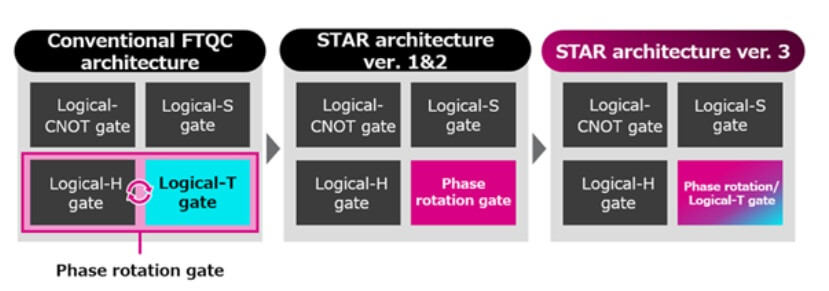
Las versiones 1 y 2 de la arquitectura STAR ya habían demostrado una mayor eficiencia en computación cuántica mediante el uso de puertas de rotación de fase únicas frente a las arquitecturas FTQC convencionales basadas en puertas T.
La versión 3 mejora la precisión computacional en más de 10 veces respecto a la versión 2 al integrar puertas de rotación de fase con puertas T lógicas.
Este avance permite realizar cálculos moleculares más complejos con el mismo número de qubits y reduce los requisitos de tasa de error de los qubits.
Figura 1 (imagen de la cabecera del artículo): Comparación de conjuntos de puertas universales en arquitecturas de computación cuántica
2.Tecnología para la optimización de modelos moleculares
Esta tecnología de optimización de modelos moleculares está diseñada para su uso en ordenadores cuánticos que implementan la arquitectura STAR versión 3 y se aplica durante el proceso de generación de circuitos cuánticos a partir de modelos moleculares.
Esta tecnología perfecciona métodos existentes, que reducen los recursos computacionales descomponiendo los modelos moleculares en múltiples términos y aplicando de forma selectiva dos técnicas —evolución temporal y muestreo aleatorio— con diferentes características según la importancia de cada término.
La técnica reconfigura el modelo molecular manteniendo la precisión de la aproximación, redistribuye la importancia de los términos y optimiza el equilibrio entre ambas técnicas. Esto minimiza el número de puertas en los circuitos cuánticos para los cálculos de energía molecular, logrando una reducción significativa del tiempo de cómputo en comparación con los métodos convencionales.
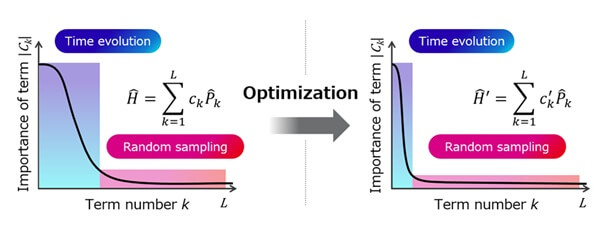
Figura 2: Principio de la optimización de modelos moleculares
Para validar la eficacia de estas tecnologías, los investigadores evaluaron el número de qubits y el tiempo de cálculo necesarios para realizar cálculos energéticos aplicables a nivel industrial en tres moléculas distintas: el citocromo P450, una enzima oxidante clave en el descubrimiento de fármacos; los clústeres de hierro-azufre, proteínas catalíticas implicadas en la síntesis de amoníaco y el metabolismo energético; y los catalizadores de rutenio, de especial relevancia en la química sintética.
En la actualidad, los cálculos energéticos precisos de estas moléculas no son viables con ordenadores clásicos debido a limitaciones de memoria. Incluso con la arquitectura STAR versión 2, estos cálculos requerirían varios milenios y sería difícil alcanzar un alto nivel de precisión debido a la escala computacional necesaria.
Los resultados de esta validación demuestran principalmente que la arquitectura STAR versión 3 reduce el número de qubits necesarios para realizar estos cálculos a entre 1/15 y 1/80 en comparación con las arquitecturas FTQC convencionales. Además, las entidades participantes confirmaron que estos cálculos son viables en ordenadores cuánticos early-FTQC incluso con una reducción en los requisitos de tasa de error físico de los qubits, pasando del 0,01 % al 0,10 %.
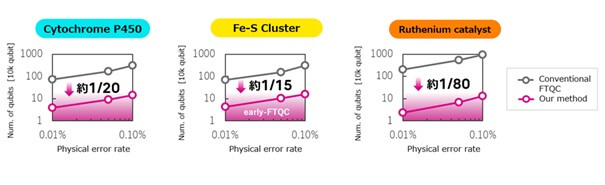
Figura 3: Número de qubits necesarios para el cálculo energético de tres moléculas
Además, la tecnología de optimización de modelos moleculares ha permitido reducir el tiempo de cálculo en tres órdenes de magnitud en comparación con su no utilización. Fujitsu y la Universidad de Osaka han confirmado que los tiempos de computación pueden reducirse de forma significativa hasta aproximadamente 35 días con una tasa de error de qubits del 0,10 % y hasta alrededor de 10 días con una tasa del 0,01 %.
Se espera que futuras reducciones en las tasas de error físico de los ordenadores cuánticos, junto con el uso de computación paralela mediante múltiples sistemas cuánticos, permitan acortar aún más los tiempos de cálculo, situándolos en niveles suficientemente prácticos.
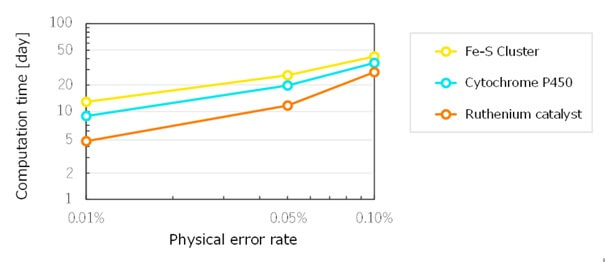
Figura 4: Tiempo de computación necesario para el cálculo energético de tres moléculas
Planes futuros
Fujitsu y la Universidad de Osaka continuarán avanzando en la arquitectura STAR y en la tecnología de optimización de modelos moleculares, ampliando el rango de aplicaciones prácticas de los ordenadores cuánticos en la era early-FTQC. Ambas entidades aspiran a contribuir a la resolución de desafíos sociales mediante la aplicación de estas tecnologías en diversos sectores industriales, como el descubrimiento de fármacos, el desarrollo de nuevos materiales y las finanzas.






